Einblicke in das tiefste Innere von lebenden Zellen
Forschungsbericht (importiert) 2018 - Max-Planck-Institut für terrestrische Mikrobiologie
Subzelluläre Strukturen sichtbar machen

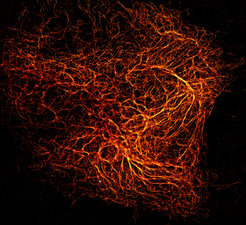
Vieles, was wir heutzutage über Lebensprozesse wissen, verdanken wir direkten Beobachtungen unter dem Mikroskop, denn mithilfe von moderner Lichtmikroskopie können wir Strukturen bis zu 200 bis 300 Nanometer (nm) auflösen und so die Vorgänge des, mit bloßem Auge nicht wahrnehmbaren, zellulären Lebens sichtbar machen. Kleinere Details, etwa einzelne Proteine oder subzelluläre Komplexe, können so jedoch nicht auseinandergehalten werden. Die Entwicklung hochauflösender Mikroskopie-Techniken, die 2014 mit dem Nobelpreis gewürdigt wurde, hat das altbewährte Feld der Lichtmikroskopie daher kräftig aufgemischt. Mit ihrer Hilfe sind nun Auflösungen von wenigen Nanometern möglich und einzelne Moleküle in lebenden Zellen und ihre Interaktionen miteinander klar sicht- und unterscheidbar [1]. Wir können sowohl den molekularen Aufbau subzellulärer Strukturen untersuchen (Abb. 1) als auch sehr schnelle, dynamische Prozesse in der Zelle direkt verfolgen.
Sich selbst färbende Zellen
Allen hochauflösenden Mikroskopietechniken ist gemein, dass die Moleküle der Zelle nicht direkt beobachtet, sondern mit Fluoreszenzfarbstoffen angefärbt und künstlich zum Leuchten gebracht werden. So sind allerdings nur jene Moleküle sichtbar, die auch ein Fluorophor tragen. Folglich müssen biologische Strukturen möglichst dicht und vollständig markiert werden [1].
Dank fluoreszierender Proteine, wie beispielsweise dem grün fluoreszierenden Protein (GFP), dessen Gen vor fast dreißig Jahren aus der Quallenspezies Aequorea victoria isoliert wurde, sind heute Beobachtungen in lebenden Zellen zur Routine geworden. Durch ihre autokatalytische Faltung können sie als unkomplizierte genetische Marker in den verschiedensten Organismen an fast jedes beliebige Zielprotein fusioniert werden. Bei der Synthetisierung des Zielproteins in der Zelle wird dieses durch das gekoppelte fluoreszierende Protein automatisch fluoreszenzmarkiert – die Zelle färbt die gewünschten Zellbestandteile somit selbst an. Diese Markierungsstrategie ist in der hochauflösenden Mikroskopie besonders aufgrund ihrer hohen Spezifität und Effizienz beliebt: Jedes Zielprotein trägt automatisch genau einen Marker. Auch tritt kein unspezifischer Hintergrund durch ungebundene Farbstoffe auf, wie er häufig bei externen Färbungen vorhanden ist. Fluoreszierende Proteine ermöglichen daher stöchiometrische, hochaufgelöste und, da zusätzliche Färbeschritte entfallen, unmittelbare Lebendzellmessungen [1].
Schonende Lebendzellmikroskopie
Ein Schlüsselmechanismus der hochauflösenden Einzelmolekülmikroskopie ist das gezielte Ein- und Ausschalten einzelner Fluorophore. Möglich ist dies, unter anderem, durch photoschaltbare fluoreszierende Proteine, die ihre optischen Eigenschaften unter Bestrahlung mit Licht einer bestimmten Wellenlänge verändern. In der Praxis sind, wegen ihrer Helligkeit und Photostabilität, besonders grün-zu-rot photokonvertierbare Proteine beliebt. Diese mussten jedoch bislang, wie für photoschaltbare Proteine typisch, mittels UV-Licht geschaltet werden, das aber schon nach kurzer Zeit massive Schäden in den zu untersuchenden Zellen verursachen kann.
In einer aktuellen Studie konnten wir einen allgemeinen Mechanismus zur Erzeugung von Varianten grün-zu-rot konvertierbarer Proteine beschreiben, die auch mit langwelligerem und damit energieärmerem Licht mittels der sogenannten primed photoconversion (PC) [2] geschaltet werden können [3]. Unsere neuen Fluoreszenzmarker können so verhindern, dass, anstatt des natürlichen Verhaltens einer Zelle, durch Lichtstress bedingte Schutz- oder Notreaktionen und somit Artefakte beobachtet werden. Bei ihrer Entwicklung machten wir uns zunutze, dass alle grün-zu-rot konvertierbaren Proteine genetisch miteinander verwandt sind und somit eine sehr ähnliche Chromophorstruktur mit ähnlichen, im Protein zueinander benachbarten Aminosäuren besitzen. Durch einen systematischen Sequenz- und Strukturvergleich konnten wir Schlüsselaminosäuren identifizieren und ihre Bedeutung für die photophysikalischen Eigenschaften der Proteine ermitteln. Wir konnten zeigen, dass eine einzelne Aminosäure, Threonin an Position 69, die PC-Photokonvertierbarkeit mittels blauem und komplementärem nah-infraroten Licht bestimmt. Durch eine einfache Punktmutation an dieser Position kann dieser Konvertierungsmechanismus somit bei allen grün-zu-rot konvertierbaren Proteinen gezielt erschlossen oder blockiert werden [3].
Mehrfarbigkeit mit gleichfarbigen Fluorophoren
Darauf aufbauend entwickelten wir eine neuartige, robuste Mehrfarbenstrategie [4]. Die gleichzeitige Aufnahme unterscheidbarer, meist verschiedenfarbiger, Fluorophore in derselben Zelle stößt in der Einzelmolekülmikroskopie lebender Zellen schnell an ihre Grenzen. Die ohnehin hohen Ansprüche, die diese Methode an einzelne Farbstoffe stellt, potenzieren sich und so bleiben meist nur wenige, suboptimale Kombinationen übrig. Durch die Kombination von einem PC-photokonvertierbaren mit einem UV-photoaktivierbaren Protein derselben Farbe können zwei Zielproteine aberrationsfrei und unabhängig voneinander mit den orthogonalen PC- und UV-Aktivierungsmechanismen aufgenommen werden. So ergibt sich ein zuverlässiges, für lebende Zellen geeignetes Bildgebungsverfahren, das allgemein anwendbar ist und von uns für Untersuchungen an Bakterien, Hefen und Säugetierzellen etabliert wurde.
Vierdimensionale Aufnahmen von Multi-Protein-Komplexen

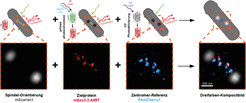
Mithilfe der Entwicklung unserer Mehrfarbenstrategie können wir dreifarbige, hochaufgelöste Bilder des Kinetochors von Schizosaccharomyces pombe aufnehmen (Abb. 2). Dieser etwa 200 nm große Multi-Protein-Komplex spielt eine entscheidende Rolle bei der Zellteilung, indem er die Mikrotubuli der mitotischen Spindel mit der DNA der Chromosomen verbindet.
Wir bestimmen zunächst die Orientierung der Kinetochorkomplexe mithilfe eines herkömmlichen, rot fluoreszierenden Referenzproteins, das die Spindelpole markiert. Danach wird das zu untersuchende Protein mittels PC und anschließend ein weiteres Referenzprotein im Zentromer mittels UV-Photoaktivierung aufgenommen. Durch die sequentielle Aufnahme aller rund 50 verschiedenen Proteine des Komplexes können wir nun in unseren aktuellen Arbeiten die vierdimensionale Organisation des Kinetochors rekonstruieren.