Die Entstehung neuer Erreger: Erkenntnisse aus dem Vergleich der Genomsequenzen pflanzenpathogener Pilze
Forschungsbericht (importiert) 2010 - Max-Planck-Institut für terrestrische Mikrobiologie
Einleitung
Krankheitserreger stellen für Landwirte eine Herausforderung dar, seit die ersten Nutzpflanzen während des Übergangs zur Agrikultur, der weltweit vor 2.000 bis 12.000 Jahren stattfand, domestiziert wurden. Obwohl krankheitserregende Organismen stets und immer während eine beträchtliche Auswirkung auf die Produktion und den Ernteertrag von Nutzpflanzen haben, wissen wir nur sehr wenig über den Ursprung und die Evolution von Krankheitserregern bei Pflanzen. Wie kommt es zum Auftreten neuer Arten von Erregern, und wie schnell können sie sich an eine neue Umgebung und neue beziehungsweise neu gezüchtete Arten oder Sorten von Wirtspflanzen anpassen? Im Vergleich zu den großen, vergangenen Zeiträumen, die die Evolution der biochemischen Maschinerie, die den Angriff der Erreger und die Verteidigung der Pflanze steuert, gebraucht hat, ist die durch den Menschen betriebene Landwirtschaft noch sehr jung. Die Domestikation von Pflanzen als Nahrungsgrundlage, zusammen mit der parallel erfolgenden Anpassung von deren Krankheitserregern, verläuft erst seit wenigen tausend Jahren.
Die Genomsequenzierung und vergleichbare genetische Methoden bieten neue Werkzeuge für das Studium der evolutionären Prozesse bei pflanzenpathogenen Pilzen. Durch den Vergleich vollständiger Genomsequenzen nahe verwandter Arten, die auf verschiedenen Wirtspflanzen in unterschiedlichen Umgebungen vorkommen, können Gene und Genabschnitte identifiziert werden, die bei deren divergierenden Entwicklung eine entscheidende Rolle gespielt haben. Die Anwendung unterschiedlicher Untersuchungsmethoden auf einen Genomdatensatz erlaubt zusätzlich Schlussfolgerungen über die Evolution der jeweiligen kompletten Genome und des Phänotyps als Reaktion auf den Übergang zur Landwirtschaft.
Der den Weizen befallende Pilzerreger Mycosphaerella graminicola als Modell für das Studium der Entstehung neuartiger Pflanzenkrankheiten
Mycosphaerella graminicola ist ein häufig vorkommender und auf Weizen spezialisierter Erreger. Er trat während der Domestikation der Wirtspflanze vor rund 11.000 Jahren im Mittleren Osten auf [1]. Die Artbildung des Pilzes ging einher mit der Wandlung seiner Wirtspflanze vom Wildgras zum Weizen, auf die eine schnelle Spezialisierung des Erregers folgte. Der Pilz hat sich seitdem weltweit überall dorthin verbreitet, wo Weizen angebaut wird.
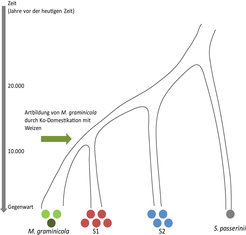
Im Mittleren Osten kann man noch heute den dort heimischen Vorläufer von Mycosphaerella auf Wildgräsern finden. Die beiden am nächsten verwandten Arten wurden Septoria 1 (S1) und Septoria 2 (S2) genannt. Obwohl die Aufspaltung dieser und der auf Weizen spezialisierten Arten erst vor kurzer Zeit erfolgte, haben sie sich infolge unterschiedlicher Wirtspflanzenarten und Umweltfaktoren seither deutlich auseinander entwickelt (Abb. 1). Impfexperimente an unterschiedlichen Grasarten mit M. graminicola (S1 und S2) zeigen, dass der Pilz sich auf Weizen spezialisiert und sich außerdem, im Vergleich zu seinen Vorgängern, zu einem virulenteren Erreger entwickelt hat. S1 und S2 sind weniger aggressive Erreger, verfügen jedoch über ein breiteres Wirtsspektrum. Diese phänotypischen Unterschiede spiegeln die selektiven Bedingungen wider, unter denen sie sich entwickelt haben. Für ein Pflanzenpathogen unterschieden sich nämlich die von einem landwirtschaftlichen Ökosystem für eine natürliche Auslese herrschenden Bedingungen sehr stark von denen eines naturbelassenen, wilden Graslandes [2]. Ein Weizenfeld ist eine dichte Population von Wirtsorganismen in Form von genetisch nahezu identischen Pflanzen, die eine schnelle Übertragung spezialisierter Erreger erlaubt. Ein Grasland besteht hingegen aus genetisch sehr unterschiedlichen Populationen verschiedener, zufällig verteilter Wirtspflanzen,, die die Evolution weniger spezialisierter Erreger begünstigen, sodass diese über die Fähigkeit verfügen, mehr als nur eine Wirtspflanze zu infizieren.
. Der Artbildungszeitpunkt von M. graminicola vor 11000 Jahren ist durch den grünen Pfeil markiert. Farbige Kreise markieren die vollständigen Genomsequenzen jeder einzelnen Art, die den populationsgenomischen Analysen zugrunde lagen. Bei der außerhalb der Gruppe liegenden Pilzart Septoria passerinii handelt es sich um ein entfernter verwandtes Gerstenpathogen.")
Analysen von Genomsequenzen erlauben Einblicke in die molekulare Basis der Spezialisierung des Erregers auf die jeweilige Wirts- beziehungsweise Nutzpflanze sowie darüber, wie diese Vorgänge durch natürliche Auslese vermittelt wurden. Durch den Vergleich der Genome der domestizierten und der wilden Erreger ist es möglich, diejenigen spezifischen Gene und Genombereiche zu identifizieren, die bei der divergierenden Entwicklung der Arten eine entscheidende Rolle gespielt haben. Der Grund dafür ist, dass die natürliche Auslese die Genomsequenzen bei den verschiedenen Erregertypen und Arten so verändert hat, dass diese in ihren speziellen Umgebungen erfolgreicher geworden sind; genau diese Unterschiede können in den Genomen festgestellt werden.
Eine weitere interessante Frage, auf die anhand der gewonnenen Datensätze eingegangen werden kann, lautet, ob bezüglich der „Evolutionsrate“ der pathogenen Erreger (das heißt der Häufigkeit, mit der sie neue Mutationen - seien sie begünstigend oder schädlich - akkumulieren) Unterschiede bestehen zwischen landwirtschaftlichen Systemen und natürlichem Grasland. Dazu wurde die Hypothese formuliert, dass der starke gerichtete Selektionsdruck, den ein landwirtschaftliches System ausübt, die Evolutionsrate eines Erregers erhöht [2]. Für einen pathogenen Pilz wurde diese Vermutung bisher noch nicht getestet.
Erstellen eines populationsgenetischen Datensatzes
Zwölf Isolate von Pilzen, die alle aus derselben Region im Norden Irans stammen, wurden für eine vollständige Genomsequenzierung ausgewählt. Diese Isolate gehörten zu den folgenden drei verschiedenen Arten: M. graminicola, gesammelt vom Weizen, und S1 und S2 von den drei Grasarten Lolium perenne, Dactylis glomerata und Elmys repens. Mithilfe neuester Sequenzierungstechnologie wurden die Genome der 12 Isolate insgesamt etwa 45-fach abgedeckt, dies bedeutet, dass jedes Basenpaar - etwa 35 Millionen je Genom - durchschnittlich 45-mal sequenziert wurde. Mithilfe umfangreicher bioinformatischer Algorhithmen wurden die 12 Genome anhand des Referenzgenoms von M. graminicola abgeglichen und analysiert. Etwa 10.500 Gene wurden so, auf 21 Chromosomen verteilt, definiert. Der vollständige Abgleich der 12 Genome umfasste etwa 9.000 Gene, deren Variation innerhalb und zwischen den Arten verglichen wurde
Genomplastizität – das Vorhandensein oder Fehlen kleiner Chromosomen

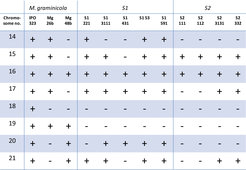
Das „Kerngenom“ des Erregers Mycosphaerella besteht aus 13 Chromosomen, die sich in den drei Arten strukturell erhalten haben. Die Genome besitzen jedoch noch zusätzlich eine hohe Anzahl kleiner Chromosomen, die in den verschiedenen Isolaten vorhanden sind oder fehlen [3]. Das Vorhandensein der kleinen Chromosomen scheint sich auf den Phänotyp des Pilzes nicht auszuwirken, wenn er in einer axenischen Kultur, also in Abwesenheit anderer Organismen, wächst. Es gibt sogar Isolate, die nur über die 13 unverzichtbaren und keines der entbehrlichen Chromosomen verfügen. Es war eine interessante Entdeckung, dass sechs beziehungsweise vier dieser Chromosomen in S1 und S2 ebenfalls vorhanden sind (Abb. 2) und dass die kleinen Chromosomen in S1 und S2 ebenso entbehrlich sind, was durch eine elektrophoretische Trennung der kleinen Chromosomen gezeigt werden konnte [4]. Die kleinen Chromosomen sind in gewissem Umfang den entbehrlichen Chromosomen von M. graminicola homolog, jedoch hat eine umfangreiche Neuanordnung die DNA-Sequenzen auf den kleinen Chromosomen durcheinander gebracht (Abb. 3). Die Tatsache, dass S1 und S2 mehrere der kleinen Chromosomen behalten haben, belegt, dass es einen selektiven Druck für die Erhaltung der entbehrlichen Chromosomen gibt, obwohl sie in einigen Individuen scheinbar leicht verloren gegangen sind.
Zur Untersuchung des Ursprungs der entbehrlichen Chromosomen wurden verschiedene Evolutions-Modelle auf den Datensatz angewendet. Es gibt deutliche Hinweise darauf, dass die kleinen Chromosomen keinen fremden Ursprung haben, was bedeuten würde, dass sie in die Genome von Mycosphaerella nicht durch eine horizontale Übertragung von anderen Organismen eingefügt wurden. Andererseits sieht es so aus, als ob die entbehrlichen Chromosomen parallel zu den Kernchromosomen entstanden sind und daher in diesen Erregern schon sehr lange vorhanden waren – zumindest, seitdem die gemeinsamen Vorläufer von M. graminicola sowie von S1 und S2 entstanden sind.

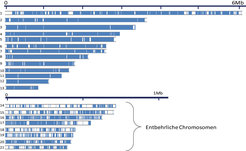
Doch wie und warum bleiben die kleinen Chromosomen in dieser Gruppe der Erreger erhalten? Eine kleine Anzahl von Genen auf den gemeinsamen entbehrlichen Chromosomen ist bei M. graminicola sowie S1 und S2 erhalten geblieben, und die Rolle genau dieser Gene könnte die treibende Kraft zur Erhaltung dieser Chromosomen in den Genomen für eine lange Zeit sein, selbst über Artgrenzen hinweg. Aktuelle Untersuchungen widmen sich der Rolle und Expression von Genen auf den entbehrlichen Chromosomen. Es kann sein, dass der Mechanismus, aufgrund dessen die Chromosomen entweder vorhanden sind oder fehlen, eine Selektion ausbalanciert: Eine balancierende, also ausgleichende Selektion tritt dann auf, wenn das Vorhandensein von Chromosomen (oder Genen) in bestimmten Situationen einen Vorteil und in anderen einen Nachteil für das Überleben bedeutet. Dieser Selektionstyp wird als Motor der Evolution einer Reihe von Genen beschrieben, die mit Pathogenität in Zusammenhang stehen. Er veranschaulicht sehr eindrucksvoll den Wettlauf zwischen Pflanzen und den sie befallenden Krankheitserregern [5]. Polymorphismen, die auf dem Vorhandensein oder Fehlen ganzer Chromosomen beruhen, sind aus anderen Pilzarten bekannt. Eine so große Anzahl von Chromosomen, wie im Fall des Erregers Mycosphaerella, ist daran jedoch sonst nie beteiligt. Anhand dieser Untersuchungen der Sequenzevolution und der Funktion der Gene auf den kleinen Chromosomen wird es möglich sein, die Rolle der Chromosomenplastizität bei Pilzerregern aufzuklären und darüber hinaus auch allgemeine Erkenntnisse über die Evolution der Genome bei Pilzen zu gewinnen.
Identifikation der an der Spezialisierung von Wirtspflanzen und der Artbildung der Erreger potenziell beteiligten Gene
Es waren vermutlich ganz bestimmte Gene, die die Anpassung der pilzlichen Erreger an die jeweilige Entstehung und durch den Menschen vorangetriebene Züchtung neuer Wirtspflanzen vorangetrieben haben. Durch eine Akkumulation adaptiver Mutationen hat sich die natürliche Selektion auf diese Gene stärker ausgewirkt. Die adaptiven Mutationen können veränderte Proteinsequenzen zur Folge haben, und diese können dann neue Funktionen ausüben, wie zum Beispiel eine veränderte Affinität zu den Genprodukten neuer oder neuartiger Wirtspflanzen. Auf ähnliche Weise können auch solche Gene betroffen gewesen sein, die beim Übergang in eine landwirtschaftliche Umgebung oder am Prozess der Artbildung selbst beteiligt waren. Um derartige positiv selektierte Gene zu identifizieren, konnte die Zahl der adaptiven Mutationen im Vergleich zu ihrer Gesamtzahl anhand verschiedener Teststatistiken und Modellierungen verglichen und analysiert werden.
Die drei Mycosphaerella-Erreger weisen nur eine sehr kleine Anzahl von Genen (etwa 20) auf, die durch eine größere Akkumulation adaptiver Mutationen verändert wurden. Interessanterweise haben sich mehrere derselben Gene mit positiv selektierten Mutationen in allen drei pathogenen Arten geändert. Dies deutet sehr darauf hin, dass diese kleine Anzahl von Genen tatsächlich an der Spezialisierung und der Artbildung beteiligt ist. Was ist die Funktion dieser Gene? Ausgehend von der Annotation des Genoms von Mycosphaerella graminicola, wurden die Gene lediglich als offene Leserahmen ohne Homologie zu anderen bekannten Genen verzeichnet. Die Gene sind wahrscheinlich nur für diese Gruppe von Gras-Pathogenen spezifisch und sind entsprechend ihrer bestimmten Funktion im Verlauf der Interaktion zwischen Erreger und Wirtspflanze entstanden.
Die Rolle dieser positiv selektierten Gene kann anhand infizierter Pflanzen charakterisiert werden. Messungen von RNA-Gehalten mittels real time PCR, die während verschiedener Stadien des Fortgangs der Erkrankung in infiziertem Gewebe vorgenommen wurden, erbrachten den Nachweis, dass drei der Kandidatengene in M. graminicola und S2 nur im Verlauf der Kolonisierung der Wirtspflanzen zu Expression kommen. Darüber hinaus zeigte eines der drei analysierten Gene im Vergleich mit einer Infektion eines Grases der Gattung Lolium eine veränderte Expression nach der Infektion von Weizen, was die Vermutung nahe legt, dass die Regulation dieses Gens von der Art der Wirtspflanze abhängt. Die Kandidatengene werden nun weiter untersucht, indem Deletionsmutanten von ihnen erzeugt werden. Mycosphaerella ist ein System, für das kürzlich brauchbare Ansätze der reversen Genetik entwickelt wurden [6]. Sobald Mutanten zur Verfügung stehen, wird deren Virulenz auf unterschiedlichen Wirtspflanzen untersucht. Um die Rolle dieser Gene, die sie bei der Affinität zur Wirtspflanze spielen, beurteilen zu können, werden die Kandidatengene in S1 und S2 übertragen, um eventuelle Änderungen der Wirtspflanzenkompatibilität zu beobachten. Diese Experimente sind noch nicht abgeschlossen.
Adaption und Evolutionsrate pathogener Pilzarten
Bekannt ist, dass domestizierte Pflanzenarten mehr unnützliche und sogar schädliche Mutationen enthalten und sich schneller verändern können als ihre undomestizierten Vorläufer [7]. Ähneln die hier vorgestellten Ergebnisse der „Ko-Domestizierung“ von M. graminicola denen von domestizierten Nutzpflanzen? Durch die Anwendung unterschiedlicher Evolutions-Modelle [8, 9] auf den Genomdatensatz von Mycosphaerella-Erregern war es möglich, den Anteil adaptiver Mutationen in den drei Arten im Verhältnis zum Anteil schädlicher Mutationen zu quantifizieren und die Evolutionsrate in den einzelnen Arten zu bestimmen. Ergebnis: Im Gegensatz zu dem sich für domestizierte Pflanzen abzeichnenden Muster weisen die „ko-domestizierten“ Erreger keine übergroße Anzahl schädlicher Mutationen auf. M. graminicola zeigt, ganz im Gegenteil, im Vergleich zu seinen wild lebenden Vorläufern ein Übermaß an vorteilhaften Mutationen. Eine Bewertung des Umfangs der Selektion zeigt zusätzlich, dass M. graminicola bei der Eliminierung schädlicher Mutationen äußerst erfolgreich ist. Die evolutionären Eigenschaften der Erreger unterscheiden sich daher deutlich von denjenigen der domestizierten Pflanzenarten. Während der „Ko-Domestizierung“ von M. graminicola selektierte die landwirtschaftliche Umgebung somit einen hochgradig wirtsspezifischen Erreger mit der Fähigkeit, sich schnell an neue Bedingungen anzupassen, wie zum Beispiel an neue Varianten und Sorten des Weizens oder sogar an Fungizide.
Ausblick
Zur weiteren Charakterisierung der funktionalen Unterschiede zwischen M. graminicola, S1 und S2 wird eine vollständige Transkriptomdatenserie erstellt. Diese Daten erlauben die Untersuchung der Genexpression des Erregers im Verlauf der Infektion und gleichzeitig der Expression der Pflanzengene als Reaktion auf die unterschiedlichen Erreger. Dies erlaubt zum Beispiel die Erkennung derjenigen Gene, die im Weizen beispielsweise durch M. graminicola, jedoch nicht durch S1 aktiviert werden, sowie die Beantwortung der Frage, welche Pathogenitätsfaktoren während der Infektion eines Wildgrases in S2, jedoch nicht in M. graminicola aktiviert werden.
Die hier vorgestellten Studien fokussieren auf die evolutionären und funktionalen Eigenschaften der entbehrlichen Chromosomen. Jedoch ist ein Verständnis der molekularen Eigenschaften, die es überhaupt zulassen, dass diese Chromosomen während der Meiose verloren gehen, ebenfalls und unbedingt erforderlich, um weitere Grundlagen von Vererbungsmechanismen zu erkennen. Mithilfe von Hochdurchsatz-Sequenzierungstechnologien (Chip Sequencing) wird nunmehr versucht, auf den entbehrlichen Chromosomen Zentromere zu identifizieren. Man vermutet, dass die kleinen Chromosomen vielleicht entweder keine Zentromere besitzen oder dass ihre Zentromere nicht korrekt funktionieren. Die Zentromerbereiche eines Chromosoms sind bei der Zellteilung entscheidend an der korrekten Aufteilung der Chromosomen auf die Tochterzellen beteiligt. Mit der Chip-Sequencing-Methode wird es möglich sein, die Bindungsproteine der Zentromere zu isolieren und die DNA-Sequenz zu entschlüsseln, an die sie sich binden. Die Charakterisierung dieser molekularen Eigenschaften ist für ein volles Verständnis des Verlorengehens oder Auftauchens dieser rätselhaften kleinen Chromosomen unerlässlich.